Reversions Complicate Phylogenies
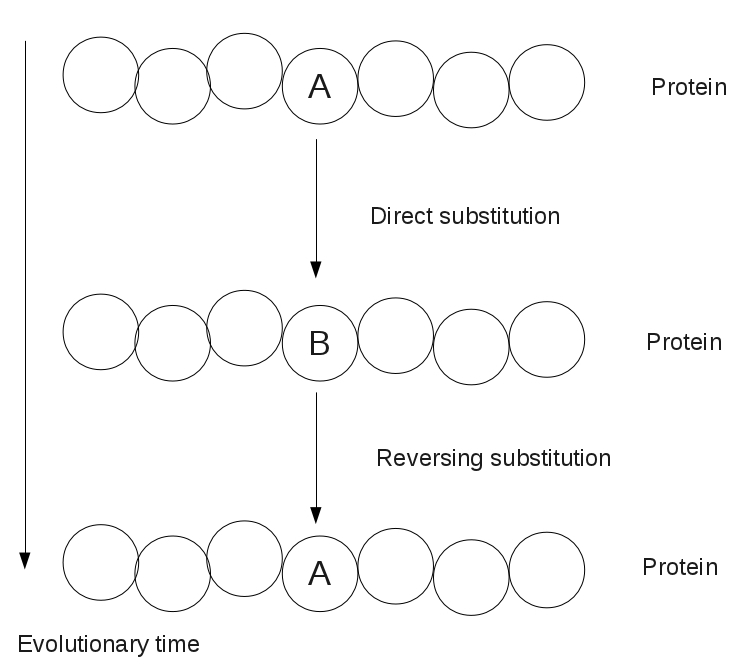
Figure 1. Illustration of an amino acid's reversing substitution after two point mutations.In “Fixing an Inconsistent Character Set”, we mentioned how the construction of a phylogeny can be complicated by a the reversion of a character to a past state. For that matter, in calculating Hamming distance and edit distance, the assumption of parsimony required us to assume that if some nucleotide base or amino acid is aligned with an identical symbol in two genetic strings, then it has not changed on the evolutionary path between the two taxa.
However, this model is too strict in practice, where a base or amino acid can change to another state and then change back as the result of two point mutations, which is called a reversing substitution; see Figure 1. In the case of DNA, the presence of only four bases makes randomly occurring reversing substitutions common; these substitutions will carry over into amino acid language via transcription and translation.
Unfortunately, with the possible exception of experimental evolution in bacteria, we lack the luxury of knowing the ancestral state of a nucleic acid strand. Instead, we must infer its most probable ancestor from homologous strands. To do so, we may use characters to construct a phylogeny (see “Character-Based Phylogeny”), then apply alignment-based phylogeny to infer strings for the tree's internal nodes (see “Alignment-Based Phylogeny”). Only once we have an adequate picture of the entire phylogeny, including its internal nodes, can we hope to identify reversing substitutions.
For a rooted tree
In other words, the third condition demands that a reversing substitution must be contiguous: no other substitutions can appear between the initial and reversing substitution.
Given: A rooted binary tree
Return: A list of all reversing substitutions in
(((ostrich,cat)rat,mouse)dog,elephant)robot; >robot AATTG >dog GGGCA >mouse AAGAC >rat GTTGT >cat GAGGC >ostrich GTGTC >elephant AATTC
dog mouse 1 A->G->A dog mouse 2 A->G->A rat ostrich 3 G->T->G rat cat 3 G->T->G dog rat 3 T->G->T