May 11, 2013, 10:34 a.m. by Rosalind Team
Topics: Alignment, Bioinformatics Tools
Jumping Genes
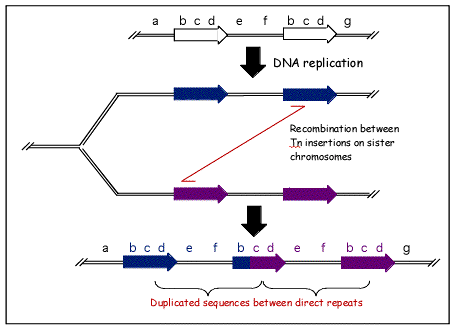
Figure 1. An illustration of a specific type of transposon called a Tn element, which recombines with a copy of itself farther down the chromosome during meiosis. As a result, the region between the transposon becomes duplicated, and an additional Tn element is formed. ©Stanley Maloy, University of California, Irvine.Different regions of the genome evolve in different ways. In coding regions, where any significant change can be lethal to the organism, the most common source of variation is point mutations. In non-coding regions (introns or intergenic spacers), we find a different situation: entire intervals can easily be duplicated, inserted, deleted, or reversed.
During the 1940s and 1950s, Barbara McClintock discovered the effect of genetic transposition in maize plants and demonstrated that many genome rearrangements are caused by regions called transposons, or relatively short intervals of DNA that can change their location within the genome. For the discovery of these "jumping genes", McClintock was awarded the Nobel prize for Physiology or Medicine in 1983.
Figure 1) illustrates the mechanism of transposon duplication during recombination. In this figure, two transposons are turned into three, and any genes located between the two original occurrences of the transposon will be duplicated. This process can also work in the opposite direction to delete genes. Both duplication and deletion can prove harmful to the organism, although deletion is more likely to be harmful.
As with any genomic region, transposons may undergo point mutations and wind up not being completely identical: they only need to be similar enough to facilitate recombination. However, you cannot find multiple repeats in the strings by using the common method for local alignment, as it will find only a single best match.
For example, if you try to locally align the strings "AAAATTTTT" and "TTTTTAAAA", the following optimal local alignment will be returned:
001 1 TTTTT 5 ||||| 002 5 TTTTT 9To find multiple similar regions in two genetic strings simultaneously, we will need to apply a technique called suboptimal alignment, which can obtain more than one different local alignment. When aligning "AAAATTTTT" and "TTTTTAAAA", we should also find the region "AAAA" of similarity:
001 6 AAAA 9 |||| 002 1 AAAA 4To obtain multiple alternative matches via suboptimal alignment, we can use the program
Lalign.Lalignhas input and output parameters similar to those ofNeedleandWater(the "+5/-4" matrix is equivalent to DNAfull), but the user can limit the number of alignments reported by changing the "expectation threshold", which is the maximum number of times a match is expected to occur by random chance. A higher expectation threshold will return more disjoint regions of similarity in the two input strings.
The Lalign program for finding multiple alternative matches via suboptimal alignment is available here.
Given: Two DNA strings
Return: The total number of occurrences of
>Rosalind_12 GACTCCTTTGTTTGCCTTAAATAGATACATATTTACTCTTGACTCTTTTGTTGGCCTTAAATAGATACATATTTGTGCGACTCCACGAGTGATTCGTA >Rosalind_37 ATGGACTCCTTTGTTTGCCTTAAATAGATACATATTCAACAAGTGTGCACTTAGCCTTGCCGACTCCTTTGTTTGCCTTAAATAGATACATATTTG
2 2
Programming Shortcut
Lalignis part of William Pearson's FASTA package, which you can download from its homepage The FASTA package manual contains descriptions of this program and its command line arguments.