Feb. 21, 2014, 5:37 p.m. by Rosalind Team
Topics: Bioinformatics Tools, NGS
All That is Said Literally
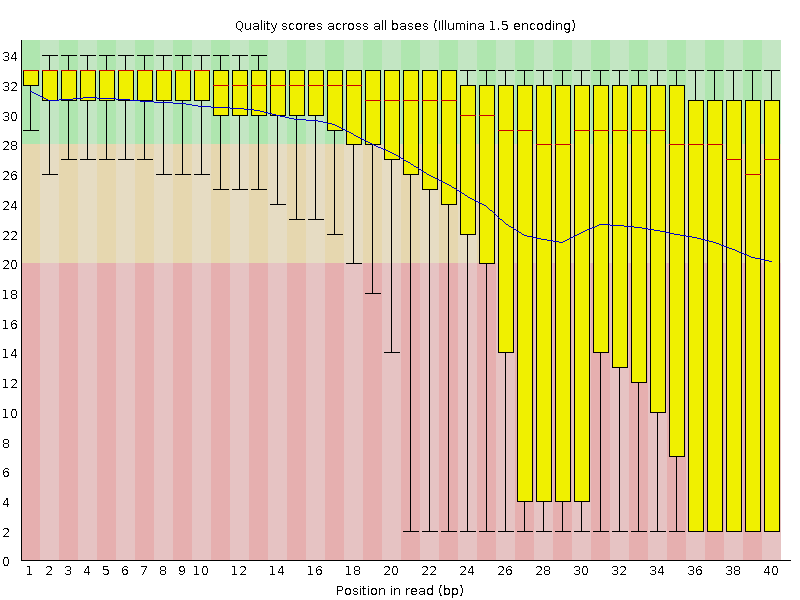
Figure 1. Per base quality scores, good dataset.Figure 2. Per base quality scores, poor dataset.Another simple quality metric for the QC is a base call quality. As often quality of reads degrades over the course of a sequence it is common practice to determine the average quality of the first, second, third,...nth base by just averaging over all reads in a file.
In the Figure 1 shown an example of a good quality dataset. Read line here is a median value, yellow box - inter-quartile range (25-75%), whiskers - 10% and 90% points. As you can see the sequence reads are 40 bases long, and median value do not fall below 37.
In the Figure 2 shown a poor dataset, and as you can see quality distribution is much wider, and average sequence quality (depicted by the blue line) is steadily declining.
In some Illumina kits the sequence quality goes up a bit first before it starts to decline, because the machine starts sequencing at lower intensity.
Quality of the bases can vary depends on position in read due to nature of the sequencing procedure. One can check this quality distribution using "Per Base Sequence Quality" module of the FastQC program.
Average accepted quality values is a 10 for the lower quartile and 25 for median. If the values falls below this limit, then the module returns a warning.
Note that for the reads >50bp long FastQC will group the bases. To show data for every base in the read use "--nogroup" option.
Given: FASTQ file, quality threshold
Return: Number of positions where mean base quality falls below given threshold
26 @Rosalind_0029 GCCCCAGGGAACCCTCCGACCGAGGATCGT + >?F?@6<C<HF?<85486B;85:8488/2/ @Rosalind_0029 TGTGATGGCTCTCTGAATGGTTCAGGCAGT + @J@H@>B9:B;<D==:<;:,<::?463-,, @Rosalind_0029 CACTCTTACTCCCTAGCCGAACTCCTTTTT + =88;99637@5,4664-65)/?4-2+)$)$ @Rosalind_0029 GATTATGATATCAGTTGGCTCCGAGAGCGT + <@BGE@8C9=B9:B<>>>7?B>7:02+33.
17
Programming Shortcuts
We can use already familiar
.letter_annotationsdictionary from “Read Quality Distribution”