July 24, 2012, midnight by Sonya Alexandrova
Topics: Computational Mass Spectrometry
Chaining the Amino Acids
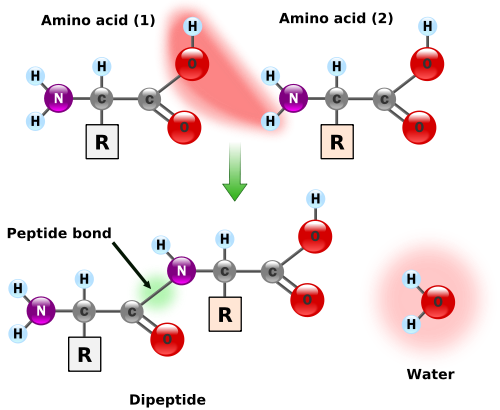
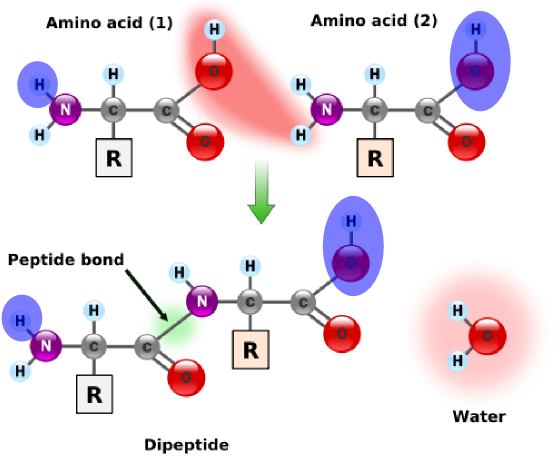
Figure 1. Formation of a peptide bondFigure 2. Outermost acidsIn “Translating RNA into Protein”, we examined the translation of RNA into an amino acid chain for the construction of a protein. When two amino acids link together, they form a peptide bond, which releases a molecule of water; see Figure 1. Thus, after a series of amino acids have been linked together into a polypeptide, every pair of adjacent amino acids has lost one molecule of water, meaning that a polypeptide containing
$n$ amino acids has had$n-1$ water molecules removed.More generally, a residue is a molecule from which a water molecule has been removed; every amino acid in a protein are residues except the leftmost and the rightmost ones. These outermost amino acids are special in that one has an "unstarted" peptide bond, and the other has an "unfinished" peptide bond. Between them, the two molecules have a single "extra" molecule of water (see the atoms marked in blue in Figure 2). Thus, the mass of a protein is the sum of masses of all its residues plus the mass of a single water molecule.
There are two standard ways of computing the mass of a residue by summing the masses of its individual atoms. Its monoisotopic mass is computed by using the principal (most abundant) isotope of each atom in the amino acid, whereas its average mass is taken by taking the average mass of each atom in the molecule (over all naturally appearing isotopes).
Many applications in proteomics rely on mass spectrometry, an analytical chemical technique used to determine the mass, elemental composition, and structure of molecules. In mass spectrometry, monoisotopic mass is used more often than average mass, and so all amino acid masses are assumed to be monoisotopic unless otherwise stated.
The standard unit used in mass spectrometry for measuring mass is the atomic mass unit, which is also called the dalton (Da) and is defined as one twelfth of the mass of a neutral atom of carbon-12. The mass of a protein is the sum of the monoisotopic masses of its amino acid residues plus the mass of a single water molecule (whose monoisotopic mass is 18.01056 Da).
In the following several problems on applications of mass spectrometry, we avoid the complication of having to distinguish between residues and non-residues by only considering peptides excised from the middle of the protein. This is a relatively safe assumption because in practice, peptide analysis is often performed in tandem mass spectrometry. In this special class of mass spectrometry, a protein is first divided into peptides, which are then broken into ions for mass analysis.
In a weighted alphabet, every symbol is assigned a positive real number called a weight. A string formed from a weighted alphabet is called a weighted string, and its weight is equal to the sum of the weights of its symbols.
The standard weight assigned to each member of the 20-symbol amino acid alphabet is the monoisotopic mass of the corresponding amino acid.
Given: A protein string
Return: The total weight of
SKADYEK
821.392