Aug. 3, 2012, midnight by Rosalind Team
Topics: String Algorithms
Identifying Unknown DNA Quickly
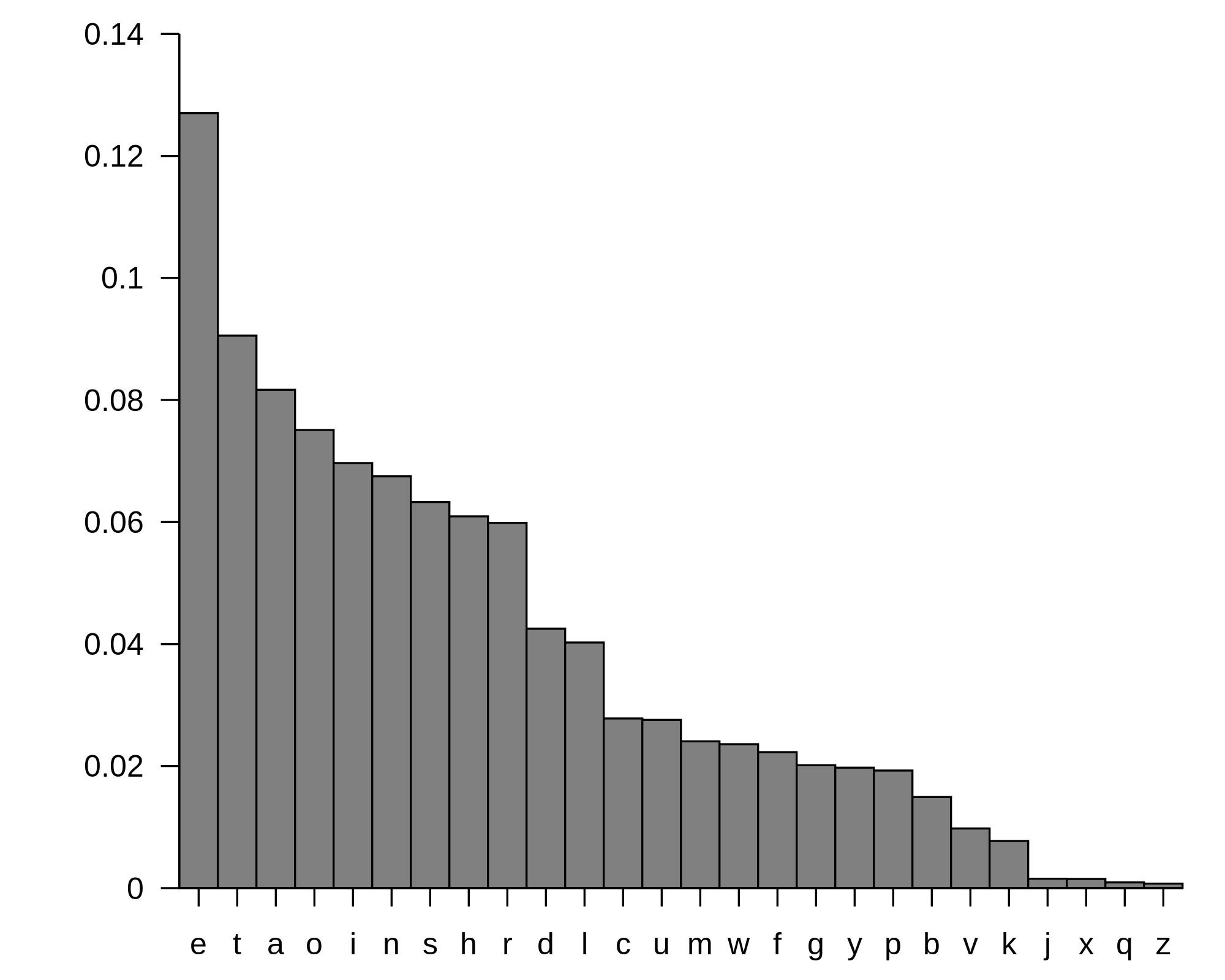
Figure 1. The table above was computed from a large number of English words and shows for any letter the frequency with which it appears in those words. These frequencies can be used to reliably identify a piece of English text and differentiate it from that of another language. Taken from http://en.wikipedia.org/wiki/File:English_letter_frequency_(frequency).svg.A quick method used by early computer software to determine the language of a given piece of text was to analyze the frequency with which each letter appeared in the text. This strategy was used because each language tends to exhibit its own letter frequencies, and as long as the text under consideration is long enough, software will correctly recognize the language quickly and with a very low error rate. See Figure 1 for a table compiling English letter frequencies.
You may ask: what in the world does this linguistic problem have to do with biology? Although two members of the same species will have different genomes, they still share the vast percentage of their DNA; notably, 99.9% of the 3.2 billion base pairs in a human genome are common to almost all humans (i.e., excluding people having major genetic defects). For this reason, biologists will speak of the human genome, meaning an average-case genome derived from a collection of individuals. Such an average case genome can be assembled for any species, a challenge that we will soon discuss.
The biological analog of identifying unknown text arises when researchers encounter a molecule of DNA from an unknown species. Because of the base pairing relations of the two DNA strands, cytosine and guanine will always appear in equal amounts in a double-stranded DNA molecule. Thus, to analyze the symbol frequencies of DNA for comparison against a database, we compute the molecule's GC-content, or the percentage of its bases that are either cytosine or guanine.
In practice, the GC-content of most eukaryotic genomes hovers around 50%. However, because genomes are so long, we may be able to distinguish species based on very small discrepancies in GC-content; furthermore, most prokaryotes have a GC-content significantly higher than 50%, so that GC-content can be used to quickly differentiate many prokaryotes and eukaryotes by using relatively small DNA samples.
The GC-content of a DNA string is given by the percentage of symbols in the string that are 'C' or 'G'. For example, the GC-content of "AGCTATAG" is 37.5%. Note that the reverse complement of any DNA string has the same GC-content.
DNA strings must be labeled when they are consolidated into a database. A commonly used method of string labeling is called FASTA format. In this format, the string is introduced by a line that begins with '>', followed by some labeling information. Subsequent lines contain the string itself; the first line to begin with '>' indicates the label of the next string.
In Rosalind's implementation, a string in FASTA format will be labeled by the ID "Rosalind_xxxx", where "xxxx" denotes a four-digit code between 0000 and 9999.
Given: At most 10 DNA strings in FASTA format (of length at most 1 kbp each).
Return: The ID of the string having the highest GC-content, followed by the GC-content of that string. Rosalind allows for a default error of 0.001 in all decimal answers unless otherwise stated; please see the note on absolute error below.
>Rosalind_6404 CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC TCCCACTAATAATTCTGAGG >Rosalind_5959 CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT ATATCCATTTGTCAGCAGACACGC >Rosalind_0808 CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC TGGGAACCTGCGGGCAGTAGGTGGAAT
Rosalind_0808 60.919540
Note on Absolute Error
We say that a number
$x$ is within an absolute error of$y$ to a correct solution if$x$ is within$y$ of the correct solution. For example, if an exact solution is 6.157892, then for$x$ to be within an absolute error of 0.001, we must have that$|x - 6.157892| < 0.001$ , or$6.156892 < x < 6.158892$ .Error bounding is a vital practical tool because of the inherent round-off error in representing decimals in a computer, where only a finite number of decimal places are allotted to any number. After being compounded over a number of operations, this round-off error can become evident. As a result, rather than testing whether two numbers are equal with
$x = z$ , you may wish to simply verify that$|x- z|$ is very small.The mathematical field of numerical analysis is devoted to rigorously studying the nature of computational approximation.