Feb. 21, 2014, 5:37 p.m. by Rosalind Team
Topics: Alignment, Bioinformatics Tools
The Odd One Out
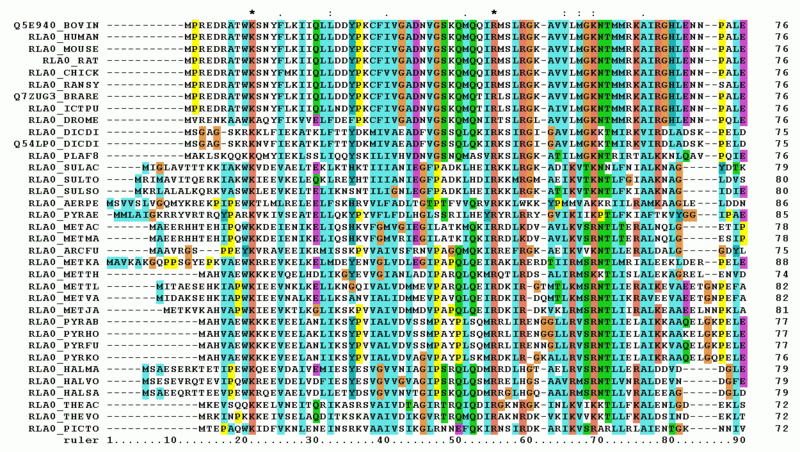
Figure 1. Fish samples waiting for analysis in the lab of US Customs and Border Protection. (Copyright: Getty Images)Figure 2. First 90 positions of a protein multiple sequence alignment of instances of the acidic ribosomal protein P0 from several organisms.Recall problems from the ABC books where you have to find out the odd object. When you're dealing with nucleotide strings you can face with the same problem - for example, if you need to detect the foreign admixture among the samples. Such analysis nowadays is preformed in the customs of the many countries in order to find the smuggling or fake food (passing tilapia off as salmon etc., see Figure 1)
In order to compare several homologous strings we need to align all of them simultaneously, a procedure known as multiple sequence alignment, or MSA. Because it requires us to compare more than two sequences at once, MSA is a more complicated problem than pairwise alignment. In fact, finding a optimal alignment between more than a very few sequences is so computationally intensive that many MSA programs rely instead on "quick and dirty" heuristic methods that are guaranteed to provide a "good" solution but not necessarily the best possible one.
See Figure 2 for an example of a multiple alignment output.
Actually MSA programs based on the series of the pairwise alignments, and optimizes sum some score over all pairs of characters in each position.
One of the first and commonly used programs for MSA is Clustal, developed by Des Higgins in 1988. The current version using the same approach is called ClustalW2, and it is embedded in many software packages. There is even a modification of ClustalW2 called ClustalX that provides a graphical user interface for MSA.
See the link below for a convenient online interface that runs Clustal on the EBI website:
Select "Protein" or "DNA", then either paste your sequence in one of the listed formats or upload an entire file.
To obtain a more accurate alignment, leave Alignment type: slow selected: if you choose
to run Clustal on only two sequences, then the parameter options correspond to those
in Needle (see “Pairwise Global Alignment”).
Given: Set of nucleotide strings in FASTA format.
Return: ID of the string most different from the others.
>Rosalind_18 GACATGTTTGTTTGCCTTAAACTCGTGGCGGCCTAGCCGTAAGTTAAG >Rosalind_23 ACTCATGTTTGTTTGCCTTAAACTCTTGGCGGCTTAGCCGTAACTTAAG >Rosalind_51 TCCTATGTTTGTTTGCCTCAAACTCTTGGCGGCCTAGCCGTAAGGTAAG >Rosalind_7 CACGTCTGTTCGCCTAAAACTTTGATTGCCGGCCTACGCTAGTTAGTTA >Rosalind_28 GGGGTCATGGCTGTTTGCCTTAAACCCTTGGCGGCCTAGCCGTAATGTTT
Rosalind_7
Programming Shortcuts
There are three main steps in the Clustal work:
Do a pairwise alignment. Program takes every pair of strings in the given set and finds the optimal global alignment for the pair constructing the distance matrix.
Create a "guide tree". Program builds the bifurcating tree using distance matrix - it takes the closest pair, adds the next closest string to that pair as a neighbour, and so on.
Use the guide tree to carry out a multiple alignment. Strings aligned progressively according to the hierarchy in the guide tree.
You can download the CLustalW2 program from its homepage and run it via the command line or using graphical interface ClustalX.
You can use installed ClustalW directly or via some wrappers:
Clustal algorithm described in details in the Clustal W and Clustal X version 2.0 paper in "Bioinformatics" journal