June 19, 2013, 7:27 a.m. by Rosalind Team
Topics: Bioinformatics Tools, NGS
How to Handle Quality
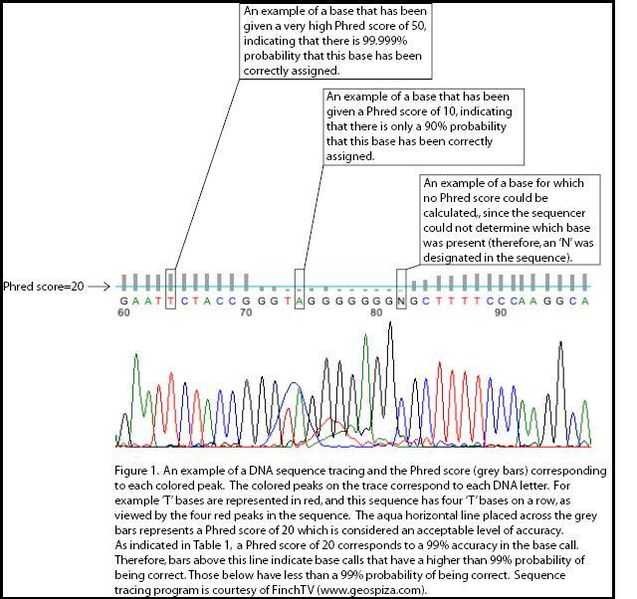
Figure 1. Phred quality scoresModern sequencing instruments generally produce a unbelievable number of reads, which are generally short sequences of <~200 bases. All sequencing machines are prone to some degree of error, so it is important to quantify the certainty with which each read is identified, or "called." This is often referred to as the quality of the base call.
This quality is customarily measured on the Phred quality scale. Lower scores indicate less confidence in the accuracy of the base call. The Phred quality score
$Q$ is calculated using the following equation:
$Q = -10\ log_{10}P$ where
$P$ is the probability that the corresponding base call is incorrect. Thus, for$Q$ =10, the probability that the base is called incorrectly is 1 in 10, and the accuracy is 90%. For$Q$ =20, the probability of error is 1 in 100 and the accuracy is 99%, and so on. See Figure 1 for an illustration.The standard format for storing the output of high throughput sequencing instruments is FASTQ format. FASTQ is an extension of FASTA format; it provides both the sequence and the per-base quality scores for each read. Nucleotides are represented by a single symbol, while Phred scores are usually 2-digit numbers. To be able to conveniently list the two together in FASTQ format, an offset of 33 is added to the score, which is then represented by the corresponding symbol from the ASCII table. Here are two examples:
- The quality of a base with a Phred score of 10 (90% accuracy) is denoted by "+" (ASCII symbol #43)
- The quality of a base with a Phred score of 40 (99.99% accuracy) is denoted by "I" (ASCII symbol #73).
The offset of 33 was chosen because the symbols 0-33 in the ASCII table are reserved for a non-printable characters, such as backspace or vertical tab.
Each record in a FASTQ file typically consists of four lines:
- A line starting with @ that contains the sequence identifier
- The actual sequence
- A line starting with + containing an optional sequence identifier or comment
- A line with quality scores encoded as ASCII symbols
Notice that the 2nd and 4th line should always have the same length.
The following is an example FASTQ record:
@HWI-ST999:102:D1N6AACXX:1:1101:1235:1936 1:N:0: ATGTCTCCTGGACCCCTCTGTGCCCAAGCTCCTCATGCATCCTCCTCAGCAACTTGTCCTGTAGCTGAGGCTCACTGACTACCAGCTGCAG + 1:DAADDDF\<B\<AGF=FGIEHCCD9DG=1E9?D>CF@HHG??B\<GEBGHCG;;CDB8==C@@>>GII@@5?A?@B>CEDCFCC:;?CCCACGenerally, a FASTQ file has the suffix .fq or .fastq.
Sometimes it's necessary to convert data from FASTQ format to FASTA format. For example, you may want to perform a BLAST search using reads in FASTQ format obtained from your brand new Illumina Genome Analyzer.
Links:
A FASTQ to FASTA converter can be accessed from the Sequence conversion website
A free GUI converter developed by BlastStation is available here for download or as an add-on to Google Chrome.
There is a FASTQ to FASTA converter in the Galaxy web platform. Note that you should register in the Galaxy and upload your file prior to using this tool.
Given: FASTQ file
Return: Corresponding FASTA records
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT + !*((((***+))%%%++)(%%%%).1***-+*****))**55CCF>>>>>>CCCCCCC65
>SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
Programming Shortcut
BioPython's Bio.SeqIO module will save the day.