July 19, 2012, midnight by Mikhail Dvorkin
Topics: Genome Assembly
Introduction to Genome Sequencing
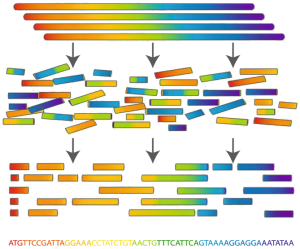
Figure 1. Fragment Assembly works by blasting many copies of the same genome into smaller, identifiable reads, which are then used to computationally assemble one copy of the genome.Recall from “Computing GC Content” that almost all humans share approximately 99.9% of the same nucleotides on the genome. Thus, if we know only a few complete genomes from the species, then we already possess an important key to unlocking the species genome.
Determining an organism's complete genome (called genome sequencing) forms a central task of bioinformatics. Unfortunately, we still don't possess the microscope technology to zoom into the nucleotide level and determine the sequence of a genome's nucleotides, one at a time. However, researchers can apply chemical methods to generate and identify much smaller snippets of DNA, called reads.
After obtaining a large collection of reads from multiple copies of the same genome, the aim is to reconstruct the desired genome from these small pieces of DNA; this process is called fragment assembly (see Figure 1).
For a collection of strings, a larger string containing every one of the smaller strings as a substring is called a superstring.
By the assumption of parsimony, a shortest possible superstring over a collection of reads serves as a candidate chromosome.
Given: At most 50 DNA strings of approximately equal length, not exceeding 1 kbp, in FASTA format (which represent reads deriving from the same strand of a single linear chromosome).
The dataset is guaranteed to satisfy the following condition: there exists a unique way to reconstruct the entire chromosome from these reads by gluing together pairs of reads that overlap by more than half their length.
Return: A shortest superstring containing all the given strings (thus corresponding to a reconstructed chromosome).
>Rosalind_56 ATTAGACCTG >Rosalind_57 CCTGCCGGAA >Rosalind_58 AGACCTGCCG >Rosalind_59 GCCGGAATAC
ATTAGACCTGCCGGAATAC
Extra Information
Although the goal of fragment assembly is to produce an entire genome, in practice it is only possible to construct several contiguous portions of each chromosome, called contigs. Furthermore, the assumption made above that reads all derive from the same strand is also practically unrealistic; in reality, researchers will not know the strand of DNA from which a given read has been sequenced.